„Ги запознаваме ретките болести“: Апертов синдром
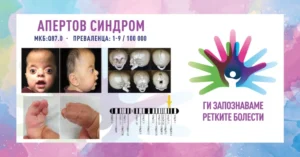
АПЕРТОВ СИНДРОМ/APERT SYNDROME
MKБ-10 Q87.0
ПРЕВАЛЕНЦА – забележана приближно кај 1/100 000 живородени деца.
ВОВЕД
Апертовиот синдром е ретко генетско нарушување кое се карактеризира со краниосиностоза (предвремена фузија на коските на черепот), абнормалности на средината на лицето и синдактилија (фузија на прстите на рацете и нозете). Состојбата е дел од групата нарушувања познати како синдроми на краниосиностоза. Апертовиот синдром обично се дијагностицира при раѓање поради неговите различни физички карактеристики и притоа се јавува потреба од мултидисциплинарна нега. Синдромот на Аперт првпат го опишал францускиот лекар Ежен Аперт, во 1906 година кога забележал уникатна комбинација на абнормалности на кранијалните нерви, лицето и рацете кај девет пациенти.
ЕТИОЛОГИЈА
Апертовиот синдром е предизвикан од мутација на генот FGFR2. Овој ген игра клучна улога во развојот и одржувањето на коските и ткивата, особено во регулирањето на растот и созревањето на клетките. Мутацијата води до абнормална сигнализација во формирањето на коските, особено во черепот и екстремитетите.
1.генетска мутација – синдромот е предизвикан од специфични мутации во генот FGFR2, кој се наоѓа на хромозомот 10; мутацијата води до предвремено спојување на кранијалните конци (краниосиностоза) и абнормален развој на коските во рацете и стапалата.
2. наследување – состојбата е обично спорадична (се јавува кај луѓе без семејна историја на нарушување), но може да се наследи и на автосомно-доминантен начин, што значи дека една копија од мутираниот ген е доволна за да ја предизвика состојбата.
3. напредна таткова возраст – постои зголемена инциденца на нови мутации кај децата родени од постари татковци, иако тоа не е единствената причина.
СИМПТОМИ И ДИЈАГНОСТИЦИРАЊЕ
Симптомите на Апертовиот синдром се разновидни и влијаат на повеќе делови од телото, првенствено черепот, лицето, рацете, стапалата, а понекогаш и на други органи. Тежината на симптомите може да варира помеѓу поединци.
1. Краниосиностоза (абнормалности на черепот)
– предвремена фузија на коските на черепот – раното спојување на кранијалните конци резултира со абнормална форма на черепот (акроцефалија или череп во облик на кула) и зголемен интракранијален притисок;
– карактеристики на лицето – поради абнормалниот развој на черепот, поединците често имаат високо чело, рамна средина и истакнати очи (проптоза) кои можат да изгледаат широко распоредени;
– хипоплазија на средното лице – неразвиеноста на средното лице може да предизвика тешкотии со дишењето и хранењето и може да доведе до опструктивна ноќна апнеа.
2. Абнормалности на рацете и стапалата
– синдактилија (спој на прсти и прсти) – една од карактеристиките на Апертовиот синдром е спојувањето на коските во прстите на рацете и нозете; вообичаено, вториот, третиот и четвртиот прст на рацете и нозете се споени;
– раце со мрежа или белезници – рацете можат да имаат мрежест изглед, а подвижноста на рацете и стапалата може да биде значително нарушена.
3. Невролошки симптоми
– задоцнување во развојот – некои лица со Апертов синдром можат да доживеат доцнење во развојните пресвртници, како што се одење и зборување;
– когнитивна функција – додека многу поединци имаат нормална интелигенција, некои можат да имаат интелектуални пречки или потешкотии во учењето, во зависност од тежината на краниосиностозата и нејзиното влијание врз развојот на мозокот.
4. Други симптоми
– проблеми со забите – вообичаени се проблеми со забите, вклучувајќи гужва, малоклузија (неусогласеност на забите) и одложено никнување на забите;
– губење на слухот – некои поединци можат да имаат спроводливо губење на слухот поради структурни абнормалности во ушите;
– абнормалности на кожата – во некои случаи, поединците можат да имаат кожни заболувања, вклучувајќи тешки акни или хиперхидроза (прекумерно потење);
– еспираторни проблеми – поради структурните абнормалности на средината на лицето, може да има опструкција на дишните патишта и тешкотии со дишењето, особено за време на спиењето (апнеа при спиење).
Дијагнозата на Апертовиот синдром се заснова на комбинација на физички преглед, студии за слики и генетско тестирање.
1. Физички преглед
– краниофацијални карактеристики – различниот изглед на черепот, лицето и рацете обично им овозможува на лекарите да се сомневаат во синдромот Аперт кратко време по раѓањето;
– абнормалности на рацете и стапалата – синдактилија на прстите на рацете и нозете е клучна дијагностичка карактеристика што помага да се разликува Апертовиот синдром од другите синдроми на краниосиностоза.
2. Студии за слики
– Х-зраци – Х-зраците на черепот, рацете и стапалата можат да помогнат да се потврди присуството на споени кранијални конци и абнормален развој на коските;
– КТ или МРИ-скенови – овие модалитети на слика се користат за да се процени степенот на краниосиностоза, да се проценат какви било абнормалности на мозокот и да се планира хируршка корекција; снимањето се користи и за следење на интракранијалниот притисок и развојот на мозокот.
3. Генетско тестирање
– тестирање на генот FGFR2 – молекуларното генетско тестирање може да ја потврди дијагнозата на Апертов синдром со идентификување на мутации во генот FGFR2; ова може да помогне да се разликува Апертовиот синдром од другите слични синдроми на краниосиностоза;
– пренатална дијагноза – во некои случаи, пренаталното генетско тестирање и снимањето со ултразвук можат да го дијагностицираат Апертовиот синдром пред раѓањето, ако постои семејна историја или ако се откријат карактеристични абнормалности при ултразвук.
ПОСЛЕДИЦИ
Со соодветна медицинска нега и хируршка интервенција, многу лица со Апертов синдром можат да водат релативно нормален живот. Очекуваниот животен век е типично нормален, иако може да има зголемен ризик од одредени компликации. Тежината на краниосиностозата, абнормалностите на лицето и синдактилијата ќе влијаат на потребата од постојана медицинска нега и операција во текот на детството и во зрелоста. Раната хируршка корекција на кранијалните и фацијалните деформитети значително ги подобрува резултатите.
ТРЕТМАН
При Апертовиот синдром, потребен е мултидисциплинарен пристап кој вклучува краниофацијални хирурзи, неврохирурзи, педијатри, специјалисти за ортопеди и други даватели на здравствени услуги. Третманот, првенствено, е фокусиран на управување со физичките абнормалности и спречување компликации.
1. Кранијална хирургија
– ремоделирање на кранијален свод – операцијата за корекција на предвременото спојување на коските на черепот обично се изведува во детството за да се олесни зголемениот интракранијален притисок, да се подобри растот на мозокот и да се нормализира обликот на черепот;
– унапредување на фронто-орбиталот – оваа операција го поместува челото и горните орбити на очите кон напред, подобрувајќи ја симетријата на лицето и спречувајќи проблеми со видот;
– напредување на средината на лицето – подоцна во детството, може да се изврши операција за да се доведе средното лице напред, да се подобри дишењето, јадењето и функцијата на говорот, како и целокупниот изглед на лицето.
2. Хирургија на раце и стапала
– синдактилско ослободување – операцијата често се изведува за да се одвојат споените прсти и прсти; ова ја подобрува функцијата на рацете, овозможувајќи подобро разбирање и фина контрола на моторот; при развојот на детето можат да бидат потребни повеќе операции;
– ортопедски менаџмент – потребна е постојана ортопедска нега за да се решат сите функционални оштетувања на рацете и нозете и да се обезбеди правилен развој на екстремитетите.
3. Ортодонтска и стоматолошка нега
– ортодонтски третман – кај поединците со Апертов синдром често има потреба од ортодонтска интервенција за да се решат стоматолошки натрупани проблеми, малоклузија и други проблеми поврзани со вилицата;
– редовна грижа за забите – раните и чести стоматолошки проценки се важни за следење на развојот на забите и решавање на какви било проблеми со забите.
4. Говорна и јазична терапија
– говорна терапија – поради хипоплазија на средното лице и можна опструкција на дишните патишта, некои лица со Апертов синдром можат да имаат потешкотии со говорот; раната интервенција со говорна и јазична терапија може да помогне во подобрување на комуникациските вештини.
5. Управување со слухот и дишењето
– слушни помагала – ако има губење на слухот, можат да бидат потребни слушни помагала за да се обезбеди правилен аудитивен развој;
– управување со апнеа при спиење – ако се дијагностицира опструктивна ноќна апнеа, потребни се третмани, како континуиран позитивен притисок на дишните патишта (CPAP) или хируршка интервенција.
6. Психолошка поддршка и дефектологија
– развојна поддршка – децата со доцнење во развојот можат да имаат корист од програмите за рана интервенција, од специјализираните образовни услуги и индивидуализирана поддршка за максимизирање на нивниот когнитивен и социјален развој;
– семејно советување – генетското советување и психолошката поддршка за семејството се важни компоненти на сеопфатната грижа, помагајќи им на семејствата да ја разберат состојбата и да се справат со емоционалните и социјалните предизвици.






